
![]() GitHub |
GitHub | ![]() GitLab mirror | Preprint
GitLab mirror | Preprint
Overview¶
Welcome to the documentation for CABS-flex standalone 3, a unified command-line package for CABS-based protein flexibility simulations, peptide structure prediction, and flexible peptide–protein docking.
CABS-flex standalone 3 builds on the previously published CABS-flex standalone (Bioinformatics 2019) and CABS-dock standalone (Bioinformatics 2019) packages. It brings their functionality into a single Python 3-based framework for running CABS simulations, reconstructing models to all-atom representation, and analyzing structural ensembles.
The new standalone version also incorporates peptide modeling workflows developed for linear and cyclic peptide structure prediction (Briefings in Bioinformatics 2024), and follows developments introduced in the CABS-flex 3.0 web server (Nucleic Acids Research 2025).
Major workflows and shared modeling core¶
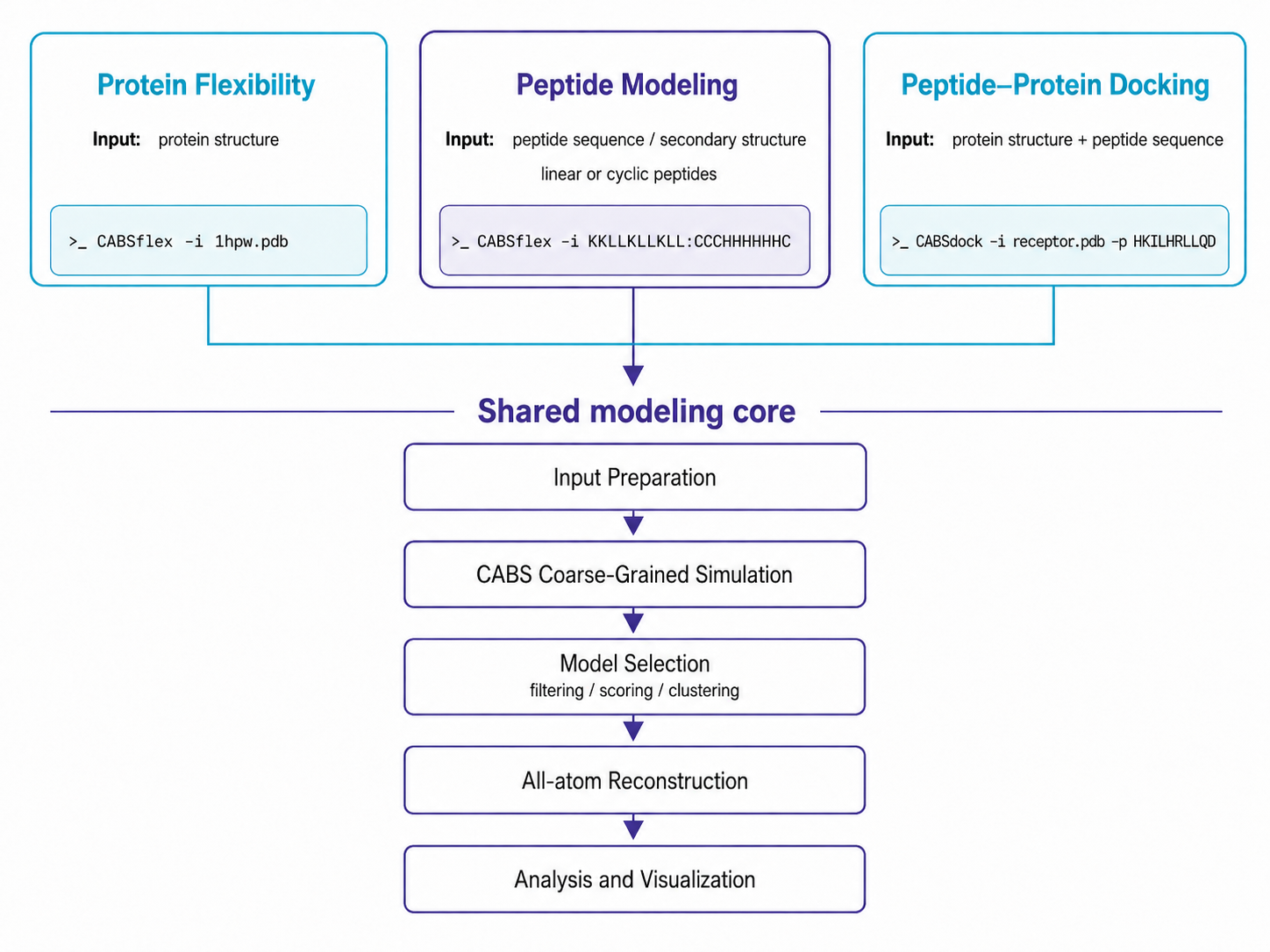
Overview of the major workflows in CABS-flex standalone 3. The package supports protein flexibility simulations, peptide structure prediction, and peptide–protein docking. All workflows share a CABS-based modeling core and, by default, return an ensemble of 10 all-atom reconstructed representative models.
Main workflows¶
CABS-flex standalone 3 supports three main workflows:
- Protein flexibility simulations: Fast simulations of protein conformational flexibility using the CABS coarse-grained model.
- Peptide structure prediction: De novo modeling of linear and cyclic peptides, including backbone-cyclized and disulfide-bonded peptides.
- Peptide–protein docking: Flexible docking of peptides to protein receptors, including cases where the binding site is unknown or receptor flexibility is important.
Quick Start¶
If you have already installed the package, you can run a quick simulation of a small protein (1hpw) with default settings:
# Activate your environment
micromamba activate cabs
# Run simulation (1hpw will be downloaded automatically)
CABSflex -i 1hpw
The simulation will take a few minutes. Results will be saved in CABSflex_output/. Check output_pdbs/model_1.pdb to model_10.pdb for the top reconstructed all-atom models.
What is new in version 3?¶
The main improvements include:
- migration from Python 2 to Python 3,
- a unified package replacing the previous standalone CABS-flex and CABS-dock distributions,
- peptide modeling workflows for linear and cyclic peptides,
- updated flexibility workflows, including pLDDT-guided restraint schemes,
- integration with cg2all for deep-learning-based all-atom reconstruction,
- improved output analysis and visualization support.
Documentation structure¶
- Start with Installation to set up the package and verify that it works.
- The Core Concepts section explains the modeling workflow, the CABS model, sampling and temperature, restraints, flexibility modes, and all-atom reconstruction.
- The User Guide section provides practical instructions for the main workflows: Protein Flexibility, Peptide Modeling, and Peptide–Protein Docking.
- The Resources section contains application examples, visualizations, and references to the related publications.
⬆ Back to top | Next: Installation