
![]() GitHub |
GitHub | ![]() GitLab mirror | Preprint
GitLab mirror | Preprint
Flexibility Modes¶
Flexibility modes are predefined recipes for generating protein restraints. They provide a simple way to control how much conformational freedom is allowed during a CABS-flex simulation. For a general explanation of restraints and custom restraint definitions, see Restraints.
How flexibility modes control restraints¶
Each flexibility mode defines how protein restraints are generated and which regions of the structure are stabilized during the simulation. For example:
flexible(ss2in older versions) generates restraints only for residue pairs where both residues belong to regular secondary-structure elements,rigid(allin older versions) generates restraints for all eligible residue pairs,plddtuses pLDDT values and secondary structure to guide restraint generation,unleashed(none/no-protein-restraintsin older versions) disables automatic protein restraint generation,ss1is an older intermediate scheme where restraints are generated when at least one residue in a pair belongs to regular secondary structure.
For custom restraints, residue-level flexibility definitions, and advanced restraint control, see Restraints.
Basic and advanced syntax¶
You can choose a flexibility mode with the -g / --protein-restraints option:
CABSflex -i protein.pdb -g flexible
Here, flexible is the selected restraint-generation mode. It generates restraints only for residue pairs where both residues belong to regular secondary-structure elements.
The full option syntax is:
-g, --protein-restraints MODE [GAP MIN MAX]
For advanced control, the same option can also be used with additional parameters:
CABSflex -i protein.pdb -g flexible 3 3.8 11.5
where:
MODEdefines the restraint-generation mode,GAPis the minimum sequence separation between restrained residues,MINis the minimum distance threshold in Å,MAXis the maximum distance threshold in Å.
In the example above:
flexibleisMODE,3isGAP,3.8isMIN,11.5isMAX.
Default restraint settings¶
The default restraint settings depend on the workflow:
| Workflow | Default setting |
|---|---|
| Protein flexibility | flexible 3 3.8 11.5 |
| Protein–peptide docking receptor | rigid 5 5.0 15.0 |
This means that standard protein flexibility simulations start from a flexibility-oriented restraint setup, while the receptor in protein–peptide docking is kept more restrained during peptide search.
Quick guide¶
| Mode | Restraint-generation recipe | When to use | Details | Older name* |
|---|---|---|---|---|
flexible |
Restraints are generated only for residue pairs where both residues belong to regular secondary-structure elements. | First-choice mode for general protein flexibility analysis. | General-purpose mode for protein flexibility. Default for the Protein flexibility workflow: flexible 3 3.8 11.5. |
ss2 |
rigid |
Restraints are generated for all eligible residue pairs according to the selected GAP, MIN, and MAX values. | Stable, well-folded globular proteins, near-native fluctuation analysis, or restrained receptor setup in docking. | Default for the protein–peptide docking receptor: rigid 5 5.0 15.0. |
all |
plddt |
Restraints are generated using pLDDT-based flexibility categories combined with secondary-structure information. | AlphaFold models or structures with available pLDDT values. | Called Rigid-pLDDT in the CABS-flex 3.0 web server. Requires pLDDT input. | |
unleashed |
No automatic protein restraints are generated. | Exploratory simulations, highly flexible systems, or large-scale conformational changes. | Sampling may be very broad and should be interpreted with caution. | none, no-protein-restraints |
ss1 |
Restraints are generated for residue pairs where at least one residue belongs to regular secondary structure. | Testing an intermediate restraint-placement scheme between flexible and rigid. |
Tested in restraint-scheme benchmarks, but not recommended as a first choice for new simulations. | ss1 |
Note
Older names are still recognized for compatibility with previous CABS-flex versions, publications, and scripts. For new runs and new documentation, use the names listed in the Mode column.
Practical recommendation¶
- Start with
flexiblefor standard protein flexibility simulations. - Use
rigidfor stable, well-folded globular proteins, near-native fluctuation analysis, or when setting up a more restrained receptor in protein–peptide docking. - Use
plddtfor AlphaFold models with meaningful pLDDT values. - Use
unleashedonly when you intentionally want to remove automatic protein restraints. - Use
ss1mainly for testing restraint-placement schemes. It may be useful as an intermediate option betweenflexibleandrigid, but benchmark tests did not support it as a first-choice mode for new simulations. -
When in doubt, compare several modes and inspect the resulting fluctuation profiles in the context of available structural or functional knowledge. A useful comparison is often:
rigidflexibleplddt, if pLDDT values are available
Detailed mode descriptions¶
Flexible mode¶
flexible mode generates protein restraints only for residue pairs where both residues are assigned to regular secondary-structure elements, such as helices and beta strands. Loop and coil regions are left unrestrained and sampled by the CABS model.
In older CABS-flex versions and publications, this restraint scheme was referred to as ss2.
This mode provides a balanced representation of protein dynamics. The underlying CABS-flex approach has been validated against crystallographic B-factors and atomistic MD simulations using various force fields in CABS-flex MD validation (JCTC 2013), and against NMR ensembles in CABS-flex NMR validation (Bioinformatics, 2014). It has also been used in structure–flexibility–function studies supported by experimental evidence, including Cryo-EM-derived conformational variability and functional analyses related to aggregation propensity and S-nitrosylation sensitivity, summarized in CABS-flex applications review (Protein Science, 2024).
Rigid mode¶
rigid mode generates restraints for all eligible residue pairs according to the selected GAP, MIN, and MAX values. As a result, it reduces fluctuations and preserves the starting conformation more strongly than flexible mode.
In older CABS-flex versions and publications, this restraint scheme was referred to as all.
This mode is useful for stable, well-folded globular proteins, near-native fluctuation analysis, or cases where large structural rearrangements are not expected. It is also used as the default receptor setup in protein–peptide docking.
In the pLDDT-restraint study, rigid-like restraint schemes showed strong agreement with MD-derived fluctuation profiles from the ATLAS database, particularly for structured, globular proteins. See pLDDT-guided flexibility modeling (Computational and Structural Biotechnology Journal, 2024).
pLDDT-guided mode¶
plddt mode uses AlphaFold pLDDT scores together with secondary-structure information to guide restraint generation. Regions with high pLDDT values are treated as more reliable and therefore more restrained, while lower-confidence regions can be allowed more flexibility.
In the CABS-flex 3.0 web server, this mode is named Rigid-pLDDT, because its overall behavior is often closer to rigid than to flexible. This mode was introduced to integrate structural confidence into CABS-flex simulations and was validated using MD simulation data from the ATLAS database. See pLDDT-guided flexibility modeling (Computational and Structural Biotechnology Journal, 2024).
pLDDT values can be provided through the B-factor column of the input PDB file or through an external .json or .tsv file.
Unleashed mode¶
unleashed mode disables automatic protein restraint generation. The conformational ensemble is then governed mainly by the intrinsic properties of the CABS coarse-grained force field and the selected simulation parameters.
This mode allows very broad sampling and can be useful for exploratory simulations, highly flexible systems, folding/unfolding-like behavior, or large-scale conformational changes. However, it may produce exaggerated motions and usually requires careful tuning of parameters such as temperature and simulation length.
The applicability of CABS-based simulations to disordered and unfolded protein systems has been reviewed in CABS-based disordered protein modeling (IJMS 2019), which discusses case studies involving disordered binding partners and unstructured regions.
SS1 mode¶
ss1 mode generates restraints when at least one residue in a pair belongs to regular secondary structure. A pair is skipped only if both residues are assigned to loop or coil regions.
This mode can be useful for testing an intermediate setup between flexible and rigid. However, in benchmark tests it was found to be less effective than the currently recommended modes, and it is therefore not suggested as a first choice for new simulations.
Example comparison of flexibility modes¶
The figure below illustrates how different restraint modes affect backbone fluctuations. The starting structure is colored by pLDDT, and the output ensembles are shown for rigid, plddt / Rigid-pLDDT, flexible, and unleashed modes.
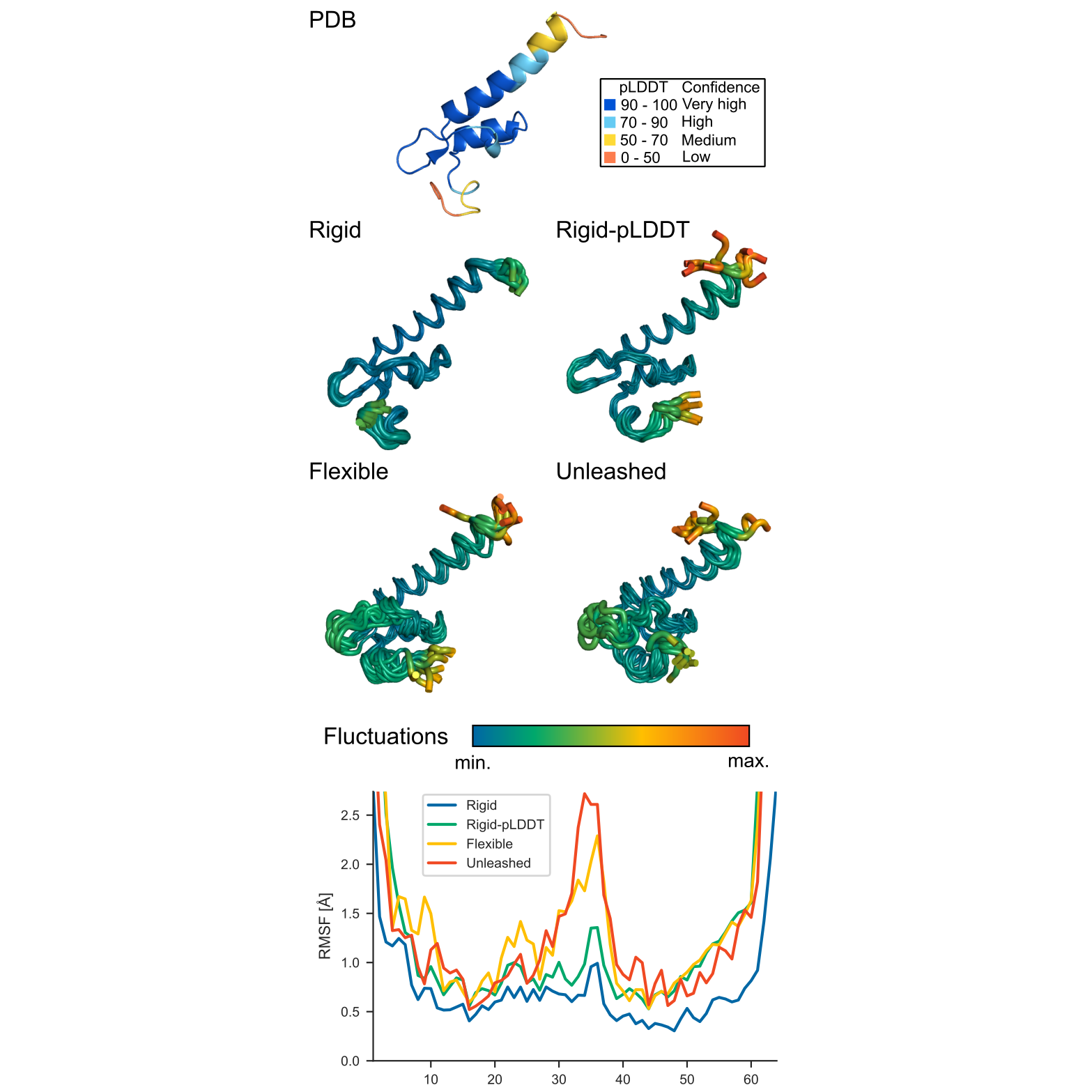
As expected, rigid gives the most restrained ensemble, flexible allows larger fluctuations in mobile regions, plddt adjusts flexibility according to structural confidence, and unleashed produces the broadest sampling.
Examples of individual modes are shown in Examples and Tutorials.
← Sampling and Temperature | ⬆ Back to top | Next: All-Atom Reconstruction