
![]() GitHub |
GitHub | ![]() GitLab mirror | Preprint
GitLab mirror | Preprint
CABS Model¶
CABS is a coarse-grained protein model designed for efficient simulations of protein structure, dynamics, folding, and interactions. The model and its applications have been reviewed in Coarse-grained protein models and their applications (Chemical Reviews 2016). CABS-based methods have been used in a broad range of applications, including protein flexibility simulations, peptide modeling, protein–peptide docking, folding simulations, loop modeling, and multiscale modeling workflows. In CABS-flex standalone 3, the CABS model provides the common simulation engine for protein flexibility, peptide modeling, and peptide–protein docking.
Representation¶
In the CABS model, each amino acid is represented by up to four pseudo-atoms:
| Symbol | Meaning |
|---|---|
| CA | C-alpha atom |
| CB | C-beta atom, except for glycine |
| SC | Center of mass of the side-chain group, except for glycine and alanine |
| cp | Center of the peptide bond |
This reduced representation preserves the key structural features needed for modeling protein flexibility, peptide conformations, and peptide–protein interactions, while enabling simulations that are orders of magnitude faster than all-atom molecular dynamics.
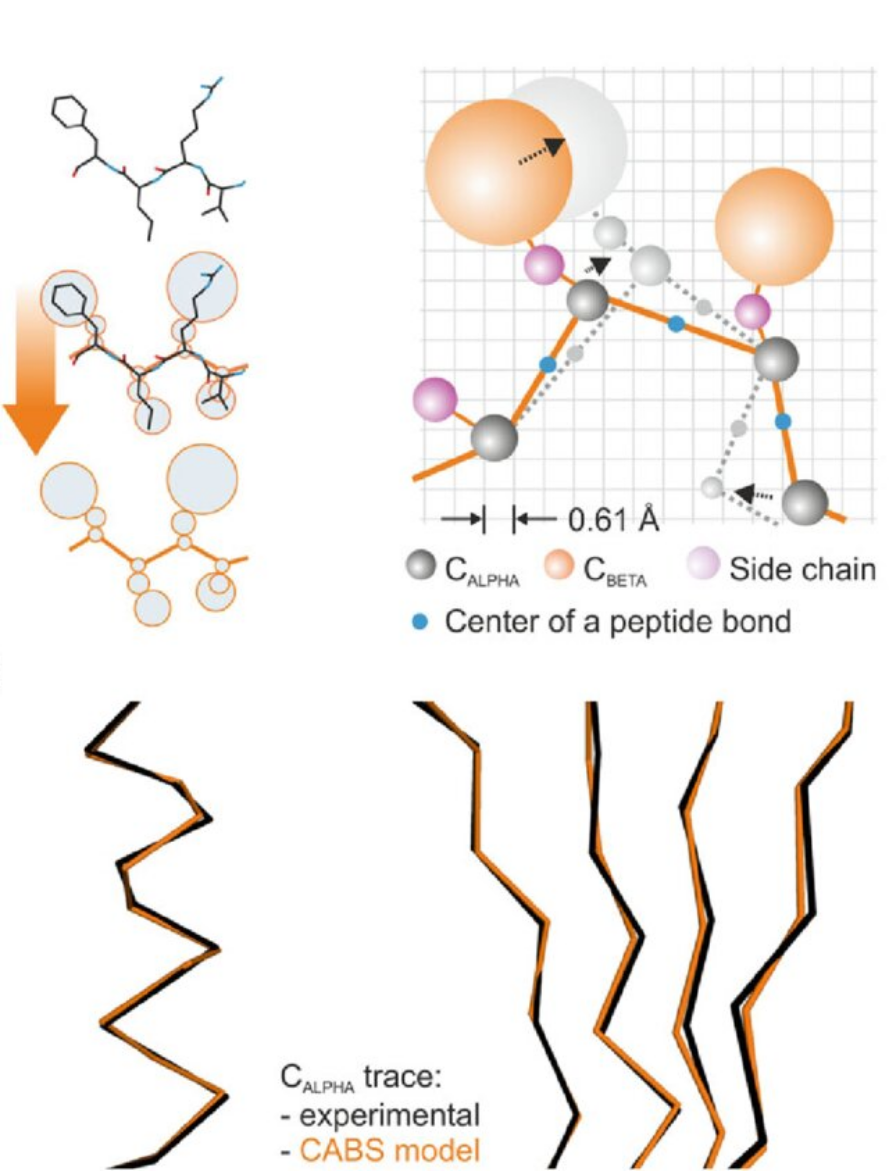 Figure. CABS coarse-grained representation.
The CABS model represents a protein chain using C-alpha, C-beta, side-chain, and peptide-bond interaction centers. The lower part of the figure illustrates how an experimental C-alpha trace is mapped onto the CABS model representation.
Figure. CABS coarse-grained representation.
The CABS model represents a protein chain using C-alpha, C-beta, side-chain, and peptide-bond interaction centers. The lower part of the figure illustrates how an experimental C-alpha trace is mapped onto the CABS model representation.
Force Field¶
The CABS force field is knowledge-based and was derived from statistical regularities observed in experimentally solved protein structures.
It includes:
- Local geometry terms: Controlling backbone stiffness and local conformational preferences.
- Statistical potentials: For side-chain interactions.
- Coarse-grained hydrogen-bonding terms.
- Implicit treatment of solvent effects.
- Optional distance restraints: Used to guide simulations.
This design allows CABS simulations to explore protein conformational changes efficiently without explicitly modeling every atom and solvent molecule.
Sampling¶
CABS simulations use Monte Carlo-based sampling to explore conformational space. Local and larger-scale conformational moves are applied to the coarse-grained representation and accepted or rejected according to the model energy.
This makes CABS suitable for efficient sampling of conformational changes that would be much more expensive to explore using classical all-atom molecular dynamics.
Efficiency¶
The coarse-grained representation makes CABS simulations substantially faster than all-atom molecular dynamics. In a benchmark comparing CABS dynamics with atomistic MD-derived fluctuation profiles, CABS dynamics was estimated to be approximately 6 × 10³ times cheaper in computational cost than classical MD for the analyzed protein set. In that comparison, a single MD simulation required on average about 3650 CPU hours, while the corresponding CABS simulation required about 0.6 CPU hour. See CABS-flex MD validation (JCTC 2013). This efficiency makes CABS-based simulations practical for rapid ensemble generation and multiscale workflows combining coarse-grained sampling with all-atom reconstruction.
Validation and applications¶
CABS-based methods have been developed and applied across several related tools and workflows.
CABS-flex uses CABS simulations to model protein flexibility and generate structural ensembles. Recent applications of CABS-flex in structure–flexibility–function studies are summarized in CABS-flex applications review (Protein Science 2024).
CABS-dock uses the CABS model for flexible peptide–protein docking, allowing conformational flexibility of both the peptide and the receptor during the search for binding poses. The CABS-dock method and its applications are described in CABS-dock flexible peptide docking (Protein Science 2020).
The CABS model has also been used for de novo modeling of linear and cyclic peptides, including workflows based on CABS-flex standalone. See linear and cyclic peptide modeling (Briefings in Bioinformatics 2024).
Further technical background¶
For a detailed technical description of the reduced representation and the original CABS model design, see Protein modeling and structure prediction with a reduced representation (Acta Biochimica Polonica 2004).
← Modeling Workflow | ⬆ Back to top | Next: Restraints