
![]() GitHub |
GitHub | ![]() GitLab mirror | Preprint
GitLab mirror | Preprint
All-Atom Reconstruction¶
CABS-flex simulations are performed in a coarse-grained representation (C-alpha trace). To make the results useful for further analysis (such as docking, binding site identification, or high-resolution refinement), the representative models are reconstructed to all-atom representation.
Default Reconstruction (cg2all)¶
By default, CABS-flex standalone 3 uses cg2all, a deep-learning-based tool for ultra-fast and accurate all-atom reconstruction from coarse-grained models. This integration ensures that the resulting ensembles are of high structural quality and suitable for immediate downstream applications.
The reconstruction process using cg2all typically handles both the backbone and side-chain atoms, providing a complete structural model.
For more information, see the cg2all publication (Structure, 2024).
Alternative Reconstruction (Modeller)¶
While cg2all is the default, CABS-flex also supports the traditional reconstruction method using Modeller. This process involves: 1. Using the C-alpha positions as constraints. 2. Optimizing the placement of backbone and side-chain atoms based on spatial restraints.
To use Modeller, you must have it installed and a valid license key configured. See Installation for details.
Cyclic and Disulfide Reconstruction (Two-Stage Hybrid)¶
When modeling cyclic peptides (using --backbone-cyclization) or structures with disulfide bridges (using --disulfide-bonds), CABS-flex employs a specialized hybrid reconstruction procedure to ensure that both coordinate accuracy and chemical topology are perfectly preserved.
Depending on your selected --aa-method, the pipeline behaves as follows:
1. Hybrid cg2all + Modeller Pipeline (Default)¶
When --aa-method cg2all is used with cyclization or disulfide constraints, CABS-flex automatically triggers a two-stage hybrid reconstruction:
- Stage 1: All-Atom Generation (cg2all): The C-alpha/coarse-grained trace is reconstructed into an initial all-atom model using
cg2all's deep-learning model to ensure high-quality sidechain and backbone placements. - Stage 2: Topological Patching (Modeller): CABS-flex immediately feeds the
cg2allall-atom structure intoModeller(viaca2allwithonly_cyclization=Trueanditerations=1). In this step:- Modeller applies the official chemical topology patches (e.g., the
DISUpatch for disulfide bridges or peptide backbone links). - It strips any excess atoms (like the
HGhydrogens in Cysteines forming a disulfide). - It performs a single, rapid iteration to physically "seal" and refine the geometry of the newly formed covalent bonds without disturbing the rest of the high-quality
cg2allprediction.
- Modeller applies the official chemical topology patches (e.g., the
Important
Modeller Requirement: Even if you choose the default cg2all method, Modeller must be installed to complete Stage 2 and successfully patch cyclic or disulfide bonds. If Modeller is missing, CABS-flex will log a warning and proceed with standard cg2all coordinates without formal covalent patching.
2. Modeller-Only Pipeline¶
When --aa-method modeller is selected, Modeller handles the entire all-atom reconstruction from scratch in a single stage, applying the standard cyclization/disulfide patches throughout its default optimization cycles.
Reconstruction Options¶
You can control the reconstruction process using several command-line flags:
Basic Control¶
-
-A, --aa-rebuild [MODE]: Enable all-atom reconstruction (default: on). The mode defines which structures to rebuild:M: rebuild Medoids (model_?.pdb) sequentially (default).C: rebuild Clusters (cluster_?.pdb) in parallel.T: rebuild Trajectories (replica_?.pdb) in batch DCD.A: rebuild ALL of the above.
-
--aa-method <ARG>: Set the method for all-atom reconstruction. Options arecg2all(default) ormodeller. -
--aa-minimize [BOOL]: Enable vacuum energy minimization of reconstructed all-atom structures using OpenMM with the modern Amber19 forcefield. Single-model medoids are minimized by default, while multi-model trajectories/replica ensembles are minimized only if explicitly requested. Passfalseto completely disable.
cg2all Specific Options¶
--cg2all-representation <REP>: Coarse-grained representation passed to cg2all.calphafor CA-only orcalpha-scfor CA plus side-chain pseudoatoms (default:calpha-sc).--cg2all-env-prefix <DIR>: Path to the isolated environment containing cg2all dependencies.
Modeller Specific Options¶
-m, --modeller-iterations <NUM>: Set the number of iterations for the reconstruction procedure in Modeller (default: 3).
Quality of Reconstruction¶
The conversion of Coarse-Grained (CG) models back to all-atom descriptions is a critical step. Recent integration with deep learning-based methods, specifically cg2all (Structure, 2024), has shown significant improvements over traditional spatial restraint methods.
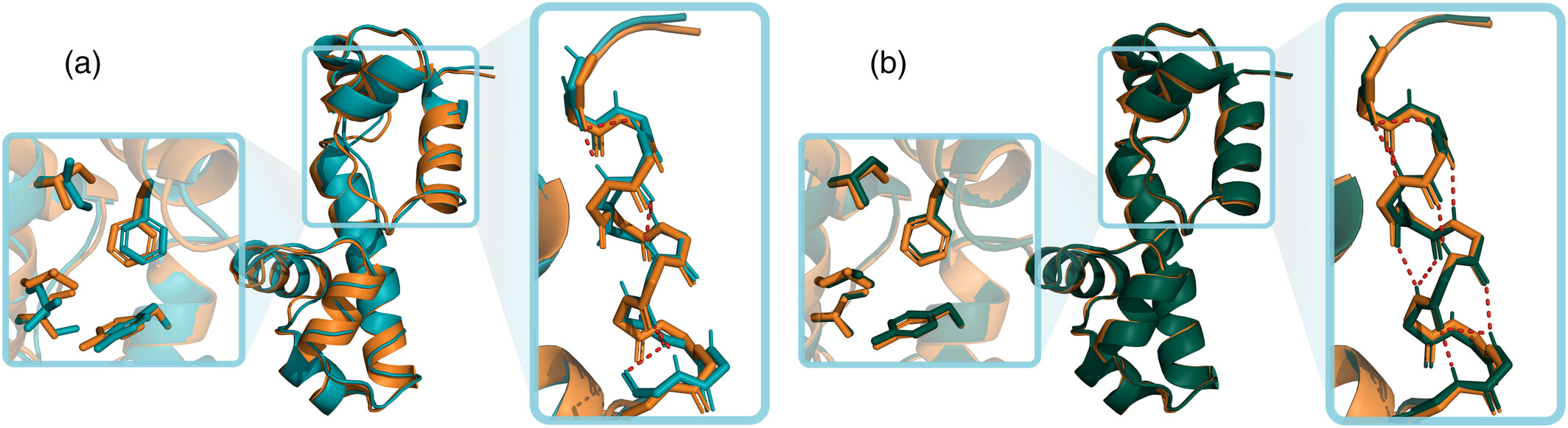 Figure: Comparison of all-atom reconstruction quality using Modeller vs. cg2all (Protein Science, 2024). The deep learning-based
Figure: Comparison of all-atom reconstruction quality using Modeller vs. cg2all (Protein Science, 2024). The deep learning-based cg2all method yields models with better physical realism, fewer steric clashes, and improved secondary structure recovery.
Energy Minimization and Refinement (OpenMM)¶
Following the all-atom reconstruction step, CABS-flex can perform an energy minimization to resolve steric clashes, improve clashscores, and relax local geometries. This refinement process is powered by the OpenMM library.
Refinement Protocol¶
- Force Field: The Amber19 force field (
amber19-all.xml) is used to model the potential energy of the protein. - Hydrogen Addition: Hydrogen atoms are automatically parameterized and added to the reconstructed heavy-atom structure.
- Environment: Minimization is performed in vacuum using the
NoCutoffnonbonded method to correct local coordinate overlaps. - Integrator: A Langevin integrator is initialized with the following parameters:
- Temperature: 300 Kelvin
- Friction Coefficient: 1.0 ps⁻¹
- Time Step: 2.0 femtoseconds (0.002 ps)
- Platform: The minimization executes on the CPU platform to ensure stability and avoid driver-level or memory limit issues when multiple reconstruction workers run concurrently.
- Optimization Limit: The energy minimization runs for a maximum of 500 iterations.
- Hydrogen Removal: After minimization, all hydrogen atoms are stripped from the output PDB. OpenMM adds hydrogens internally for energy calculations, but they are removed from the final file to keep output consistent with the heavy-atom structures produced by
cg2all.
Handling Single and Multi-Model Structures¶
- Single-Model Medoids: Single-model structures are minimized directly.
- Multi-Model Ensembles: For trajectory replica ensembles, CABS-flex processes the structures frame-by-frame, minimizing each model independently and writing them back sequentially into a multi-model PDB format.
This refinement step is enabled by default for single-model medoid reconstructions and can be toggled using the --aa-minimize flag.
← Flexibility Modes | ⬆ Back to top | Next: Protein Flexibility