
![]() GitHub |
GitHub | ![]() GitLab mirror | Preprint
GitLab mirror | Preprint
Restraints¶
Restraints are distance-based rules used to guide a CABS simulation with structural information that is considered reliable.
In a coarse-grained simulation, the CABS model and its force field determine how the system moves, samples alternative conformations, and responds to local or global structural flexibility. Restraints do not replace this sampling and do not freeze the structure. Instead, they bias the simulation by adding an energy penalty when selected distances deviate from their defined restraint distance.
In practice, restraints help preserve structural features that are expected to remain stable, while less constrained regions are left to be explored by the CABS model. This is important in protein flexibility simulations, where comparisons with all-atom molecular dynamics have shown that stabilizing selected parts of the structure can improve agreement with reference flexibility profiles. In docking simulations, restraints are used by default to keep the receptor sufficiently stable, but still flexible, during peptide search. The peptide can remain fully flexible unless specific peptide restraints are provided.
Restraints are therefore not a way to rigidly impose a single structure. They are a way to indicate which parts of the system should be trusted more strongly and which parts should be left for CABS sampling to explore.
Note
Restraints also provide a flexible way to design new restraint schemes. Such schemes can combine trusted structural information with CABS sampling: regions that are well defined can be stabilized more strongly, while uncertain, flexible, or functionally relevant regions can be left less restrained and explored more freely by the model.
How restraints work¶
Technically, a restraint adds an energy penalty when a restrained distance deviates from the defined restraint distance. Higher restraint weights make restraint-violating moves less likely to be accepted during the simulation. A distance restraint is defined between two residues. In the simplest case, it specifies:
- the first residue,
- the second residue,
- the restraint distance,
- the restraint weight.
For example:
--ca-rest-add 5:A 15:A 8.7 1.0
This means:
| Field | Meaning |
|---|---|
| 5:A | first residue: residue 5 in chain A |
| 15:A | second residue: residue 15 in chain A |
| 8.7 | restraint distance in Å |
| 1.0 | restraint weight |
The restraint distance is the reference distance used by the restraint. In this example, the restrained residue pair is expected to remain close to 8.7 Å. In the CABS restraint function shown below, no energy penalty is applied within ±1 Å of the restraint distance. For example, if the restraint distance is 8.7 Å, then no penalty is applied between approximately 7.7 Å and 9.7 Å. Outside this range, the energy penalty increases as the simulated distance becomes too short or too long. The restraint weight controls the slope of this penalty: higher weights penalize deviations more strongly.
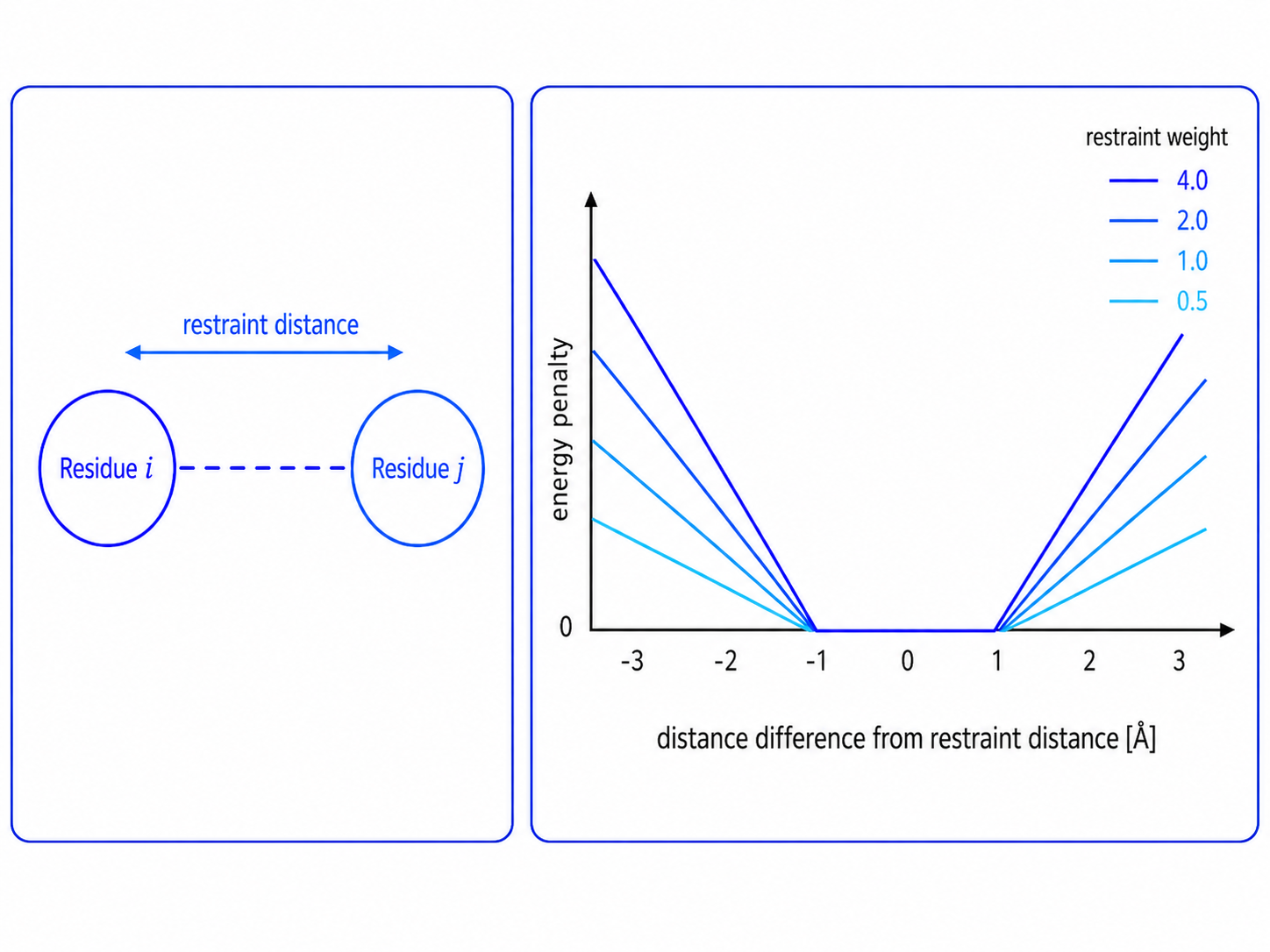
Figure. Distance restraint and energy penalty. A restraint is defined by a restraint distance and a restraint weight. In the illustrated CABS penalty function, the energy penalty is zero within ±1 Å of the restraint distance and increases outside this range. Higher restraint weights produce steeper penalty curves.
Restraints in flexibility modes¶
For protein simulations, restraints are usually not added one by one. Instead, CABS-flex can generate a complete set of protein restraints automatically according to a selected flexibility mode.
A flexibility mode defines the recipe for restraint generation: which residue pairs should be restrained, which regions should remain unconstrained by automatic protein restraints, and how the restraint scheme should be applied during the simulation. Protein restraint settings are selected with the -g option:
CABSflex -i protein.pdb -g flexible
In this example, flexible means that automatic protein restraints are generated only for residues assigned to secondary-structure elements, such as helices and beta strands. Loop and coil regions are not restrained by automatically generated protein restraints and are left to be sampled by the CABS model.
For advanced control, the -g option can also be used with additional parameters:
CABSflex -i protein.pdb -g flexible 3 3.8 11.5
where:
- flexible is the selected flexibility mode,
- 3 is the minimum sequence separation between restrained residues,
- 3.8 is the minimum distance threshold in Å,
- 11.5 is the maximum distance threshold in Å.
For guidance on choosing a mode, see Flexibility Modes.
Flexibility-mode presets¶
Flexibility modes are presets for automatic protein restraint generation.
| Mode | Restraint logic |
|---|---|
| flexible | Helices and beta strands are restrained. Loop and coil regions are left unrestricted by automatic protein restraints. |
| rigid | Automatic restraints are generated for all eligible residue pairs in the protein. |
| plddt | Automatic restraints are generated according to pLDDT-based flexibility categories combined with secondary-structure information. |
| unleashed | No automatic protein restraints are generated. |
For guidance on choosing a mode, see Flexibility Modes.
Default restraint settings¶
The default restraint settings are different for the CABS-flex and CABS-dock workflows.
| Workflow | Default setting |
|---|---|
| CABS-flex | flexible 3 3.8 11.5 |
| CABS-dock | rigid 5 5.0 15.0 |
This means that by default:
- CABS-flex uses
flexiblemode: secondary-structure elements are restrained, while loop and coil regions are not restrained by automatic protein restraints. - CABS-dock uses
rigidmode: the receptor is kept more restrained during peptide–protein docking.
The detailed meaning of flexible, rigid, plddt, and unleashed is explained on the Flexibility Modes page.
Manual restraints¶
Users can also add individual distance restraints manually.
Manual restraints are useful when specific structural information is available, such as:
- known residue–residue contacts,
- predicted contacts,
- experimental restraints,
- disulfide bonds,
- cyclic peptide constraints,
- expected peptide–protein contacts.
A single C-alpha restraint can be added with:
--ca-rest-add 5:A 15:A 8.7 1.0
Manual restraints can also be loaded from a file:
--ca-rest-file <restraint_file>.txt
For side-chain pseudoatom restraints, use:
--sc-rest-file <restraint_file>.txt
C-alpha and side-chain restraints¶
CABS-flex supports two main types of manual distance restraints:
| Restraint type | Option | Meaning |
|---|---|---|
| C-alpha restraint | --ca-rest-add, --ca-rest-file |
Restraint between C-alpha atoms of selected residues. |
| Side-chain restraint | --sc-rest-add, --sc-rest-file |
Restraint between side-chain pseudoatoms of selected residues. |
C-alpha restraints are commonly used to control backbone geometry or preserve known residue–residue distances.
Side-chain restraints are useful when the available information describes residue contacts rather than backbone positions.
Restraints in different workflows¶
| Workflow | Typical use of restraints |
|---|---|
| Protein flexibility | Control how strongly the protein follows the input structure. |
| Peptide modeling | Define cyclic peptides, disulfide bonds, or expected internal contacts. |
| Peptide–protein docking | Guide docking toward known or expected interaction regions. |
Practical recommendation¶
For standard protein flexibility simulations, start with automatic restraints through Flexibility Modes.
Use manual restraints only when you have specific structural information, such as known contacts, predicted contacts, disulfide bonds, cyclic peptide constraints, or expected peptide–protein contacts.
← CABS Model | ⬆ Back to top | Next: Sampling and Temperature