
![]() GitHub |
GitHub | ![]() GitLab mirror | Preprint
GitLab mirror | Preprint
Peptide Modeling¶
CABS-flex standalone 3 includes specialized workflows for de novo modeling of linear and cyclic peptides. The protocol uses the CABS coarse-grained protein model to efficiently sample broad conformational changes of short peptide chains, starting from sequence alone. This makes it useful for modeling flexible peptides, mini-proteins, and constrained peptide systems where extensive conformational sampling is needed.
The peptide modeling workflow is based on the multiscale protocol described in linear and cyclic peptide structure prediction (Briefings in Bioinformatics 2024). In this protocol, coarse-grained CABS-flex simulations are followed by all-atom reconstruction and optimization. The approach was evaluated for linear peptides and cyclic peptides, including backbone-cyclized peptides and peptides stabilized by disulfide bonds. Peptide modeling capabilities are also available in the CABS-flex 3.0 web server (Nucleic Acids Research 2025), while the standalone version provides command-line control over simulation settings, peptide restraints, reconstruction, and integration into larger modeling pipelines.
In CABS-flex standalone 3, peptide modeling can be used for linear peptide folding, cyclic peptide modeling, disulfide-constrained peptides, and peptide models guided by residue–residue contact information. Cyclic peptides and disulfide-constrained peptides are modeled by introducing appropriate distance restraints that close the peptide chain or enforce known covalent constraints, followed by all-atom reconstruction of representative models.
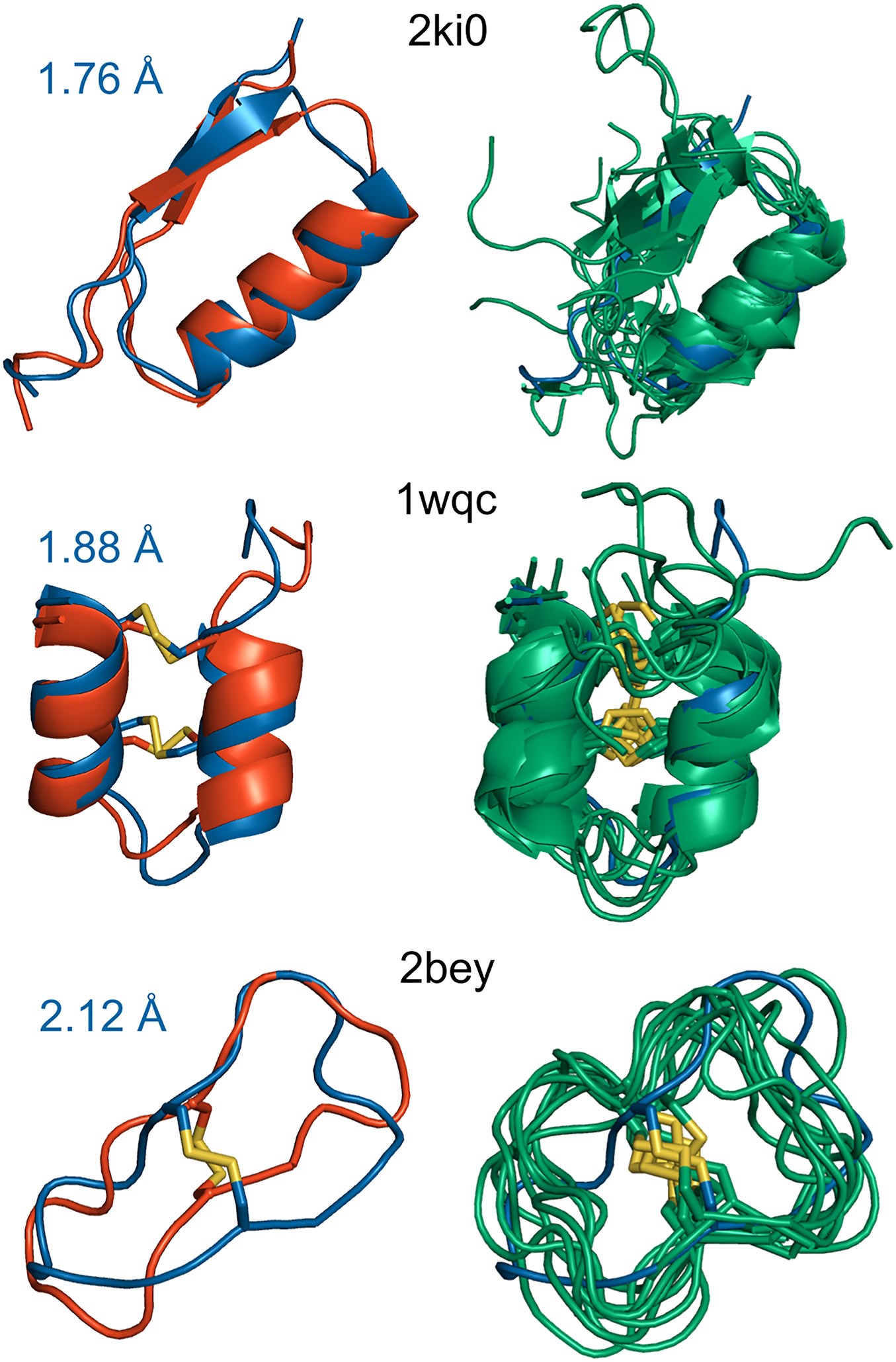
Figure. Example peptide structure predictions with CABS-flex. Predictions are shown for linear, disulfide-cyclic, and backbone-cyclic peptides. Each example includes the best model superimposed on the experimental structure and an ensemble of 10 final models, illustrating both prediction accuracy and conformational diversity.
For more information about the underlying model, see CABS Model. For information about distance restraints, see Restraints. For peptide docking directly from peptide sequence, see Peptide–Protein Docking.
Input and basic run¶
Linear Peptides¶
To model a linear peptide from its sequence, use the --peptide option:
CABSflex --peptide ACDEFGHIKLMNPQRSTVWY
This command initializes a random coil conformation and performs a simulated annealing simulation to identify the most stable folds.
Cyclic Peptides¶
Cyclic peptides are modeled by applying distance restraints between the ends of the peptide (backbone cyclization) or between specific residues (e.g., disulfide bridges or side-chain cyclization).
Key options¶
Constraints for Cyclization¶
Use --backbone-cyclization or --disulfide-bonds to define the connectivity.
Note
Peptide Chain IDs: CABSflex automatically assigns chain identifiers to peptides provided via the --peptide option in the order they appear. The first peptide is assigned PEP1, the second PEP2, and so on. You must use these identifiers when defining cyclization constraints.
-
Backbone Cyclization: Restrain the C-alpha atoms of the first and last residues. Use the chain identifier assigned to the peptide (e.g.,
PEP1).--backbone-cyclization PEP1(Enforces a default virtual C-alpha backbone restraint of 3.8 Å with a force weight of 1.0 between the first and last residues to close the peptide loop).
-
Disulfide Bonds: Restrain the side-chain pseudoatoms of specific cysteine residues. Specify the residue numbers and chain IDs.
--disulfide-bonds 3:PEP1 8:PEP1(Enforces a default side-chain pseudoatom restraint of 2.0 Å with a force weight of 1.0 between the specified Cysteine residues).
Note
Custom Cyclization: The --backbone-cyclization and --disulfide-bonds options are convenient shortcuts for standard topologies. If you need to create non-standard cyclization (e.g., linking an internal residue to a terminus, or using custom distance weights), you can still use the manual restraint flags: --ca-rest-add (for backbone) and --sc-rest-add (for side-chains).
Secondary Structure Guidance¶
You can significantly improve folding accuracy by providing predicted secondary structure information directly with the sequence, separated by a colon (:):
--peptide <sequence>:<ss_string>
For example: --peptide ACDEFGHIKLM:CHHHHCCEECC where H=helix, E=sheet, C=coil.
Advanced options¶
Sampling Intensity¶
For complex cyclic topologies or longer peptides, you may need to increase the sampling depth.
- Default Configuration: 20 annealing cycles and 50 Monte Carlo cycles.
- Tuning Options: Adjust with
--mc-annealingand--mc-cycles.
For a full guide on the simulation steps, snapshots, and how to optimize sampling depth, see the Sampling and Temperature guide.
Starting Conformation¶
If you have a starting model (e.g., from another tool), you can use it as a template:
--add-peptide <pdb_file>
Output and analysis¶
The workflow generates a trajectory of conformations which are then filtered and clustered:
- Clustering: The 1000 lowest-energy models are clustered into 10 groups.
- Representative Models: The centroids of the top 10 clusters are reconstructed to all-atom representation using cg2all.
- Files: Output models are saved in
output_pdbs/model_?.pdb.
Related pages¶
- Restraints: Detailed guide on defining cyclization and disulfide bonds.
- Peptide–Protein Docking: Using modeled peptides in docking simulations.
- Examples: Practical examples of peptide modeling.
- References: Linear and cyclic peptide modeling (Briefings in Bioinformatics 2024).
← Protein Flexibility | ⬆ Back to top | Next: Peptide–Protein Docking